
Russula nobilis, Velen.
|
publication ID |
https://doi.org/10.1016/j.phytochem.2014.08.018 |
|
DOI |
https://doi.org/10.5281/zenodo.10604732 |
|
persistent identifier |
https://treatment.plazi.org/id/03A9878C-DF17-3F42-A550-3A08FCDEFCF1 |
|
treatment provided by |
Felipe |
|
scientific name |
Russula nobilis |
| status |
|
2.2. Sesquiterpenes in damaged fruiting bodies of R. nobilis View in CoL
To study the complex patterns of secondary metabolites enzymatically formed in injured fruiting bodies, freshly collected undamaged specimens of R. nobilis were finely ground at RT without adding any solvent, then left at RT in the air; subsequently, samples of the mush were extracted with CH 2 Cl 2 at different times after injury and extracts were analyzed by TLC. Following this procedure, we observed a continuous change in the patterns of metabolites extracted in the first 30 min. Thus, to identify the compound(s) responsible for the pungency of R. nobilis to the human tongue, extraction of the mush had to be done within 30 s after crushing fruiting bodies. Separation of the extract on a RP-18 column afforded the antimicrobial dialdehyde ( —)-velleral 3, followed by unchanged velutinal esters. Velleral 3 is one of the acrid sesquiterpene dialdehydes responsible for the pungency of several Lactarius and Russula species, being formed by enzymatic conversion of velutinal esters, such as the stearate (+) — 1 ( Sterner et al., 1985; Daniewski and Vidari, 1999). Notably, velleral 3 was rapidly metabolized in damaged specimens of R. nobilis , being almost undetectable in an extract made 1 min after injury, whereas a significant amount of stearate 1 was still present. On the other hand, a TLC of an extract of R. nobilis made 20 min after mincing fruiting bodies indicated the absence of dialdehyde 3 as well as stearate 1; in their place, different more polar compounds were detected as intensely colored red–blue–green spots by staining silica gel plates with the sulphovanillin reagent. The extract was then partitioned between hexane and MeOH–H 2 O (9:1), and the components of the polar layer were subsequently separated by multiple chromatographic separations on silica gel and RP-18 columns, to give several oxygenated sesquiterpenes, namely: the known furanolactaranes furandiols (+) — 4 ( Nozoe et al., 1971; Daniewski and Vidari, 1999) and (+) — 5 ( Sterner et al., 1988; Daniewski and Vidari, 1999); the 5-lac-taranolides epoxylactone (+) — 6 ( Daniewski et al., 1993; Daniewski and Vidari, 1999), conjugated anhydrolactarorufin A 7a ( Daniewski et al., 1984; Daniewski and Vidari, 1999), and lactarorufin A (+) — 8 ( Daniewski and Kocor, 1971; Daniewski and Vidari, 1999); the 8,9-seco-5-lactaranolides blennin C ( —) — 9 ( Vidari et al., 1976; Daniewski and Vidari, 1999) and aldehydolactone ( —) — 10 ( Pang et al., 1992; Daniewski and Vidari, 1999); three new lactarane sesquiterpenoids, 12–14, named russulanobilines A–C.
NMR data indicated the structure of conjugated anhydrolactarorufin A for compound 7a, which was reported to occur in several Lactarius species ( Daniewski et al., 1984; Daniewski and Vidari, 1999); however, no physical or spectral data of an isolated sample of this sesquiterpene alcohol have been published so far ( Vidari and Vita-Finzi, 1995; Daniewski and Vidari, 1999; Clericuzio et al., 2008). Therefore, we report now the spectroscopic data of compound 7a, along with its absolute configuration. In addition to three singlets attributable to the three characteristic methyl groups of the lactarane skeleton, one of which was located on a double bond ( δH 1.93, Me-12), the 1 H NMR spectrum showed these characteristic resonances for structure 7a: an isolated olefinic proton at δH 5.93 assigned to H-4, an AB system collapsed at δH 4.85 attributable to H 2 -13, one proton doublet ( J = 10.8 Hz) at δH 4.44 assignable to H-8, trans to H-9, two multiplets at δH 2.78 and at δH 2.30 attributed to H-2 and H-9, respectively; a triplet ( J = 12.5 Hz) at δH 1.44 along with a coupled double-doublet ( J = 12.5 and 6.2 Hz) at δH 2.02, assigned to H 2 -1; a double-doublet ( J = 14.0 and 7.2 Hz) at δH 1.73, along with a coupled double-doublet ( J = 14.0 and 2.2 Hz) at δH 1.88, assignable to H 2 -10. The 13 C NMR spectrum ( Table 1 View Table 1 ) was fully consistent with structure 7a. Moreover 7a was converted by standard acetylation conditions to 7b, which was identical, including the optical rotation, with the dehydration product of the very well-known (+)-lactarorufin A - 8 ( Daniewski and Kocor, 1970; Daniewski et al., 1976).
As to the new compounds 12–14, biogenetic considerations and the presence of three characteristic methyl signals in the NMR spectra, two of which geminal to a quaternary sp 3 carbon (C-11) and the third one bonded to a methine (C-3), clearly suggested they were lactarane derivatives ( Vidari and Vita-Finzi, 1995; Daniewski and Vidari, 1999; Clericuzio et al., 2008). Moreover, a strong band at about 1740–1770 cm — 1 in the IR spectra and the signal for an ester carbonyl group at δC 170–175 ppm in the 13 C NMR spectra indicated that all the three compounds were unsaturated Ƴ- lactones.
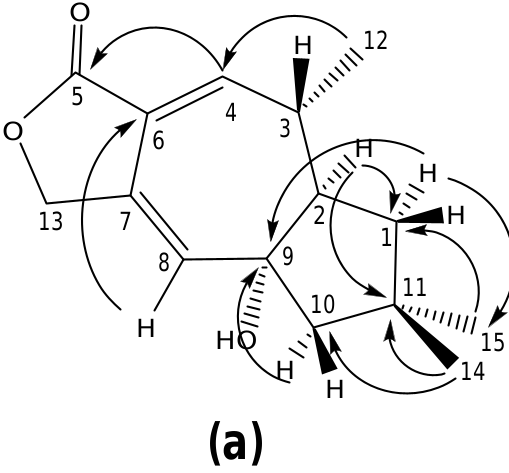
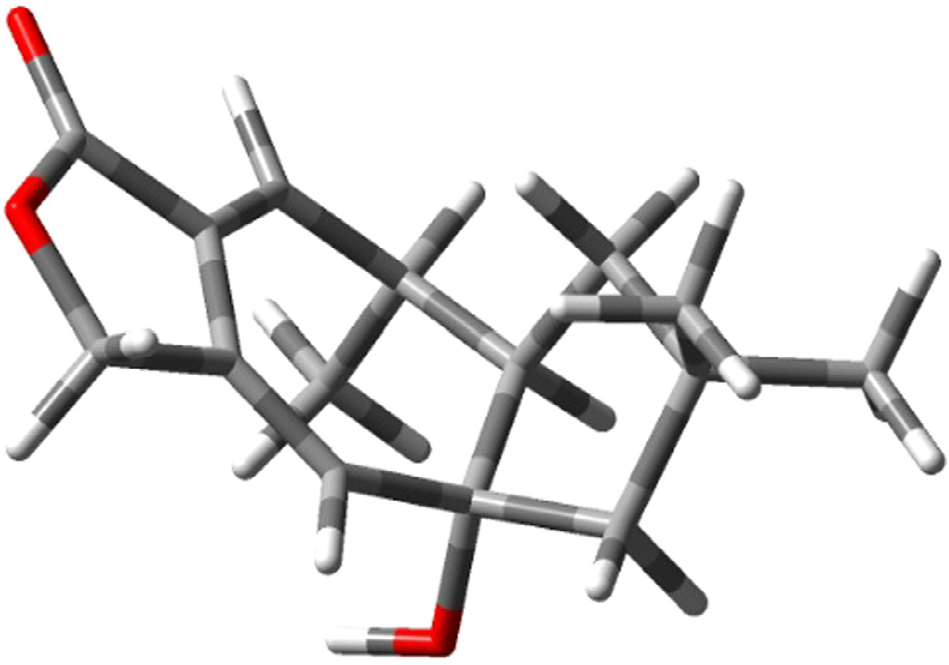
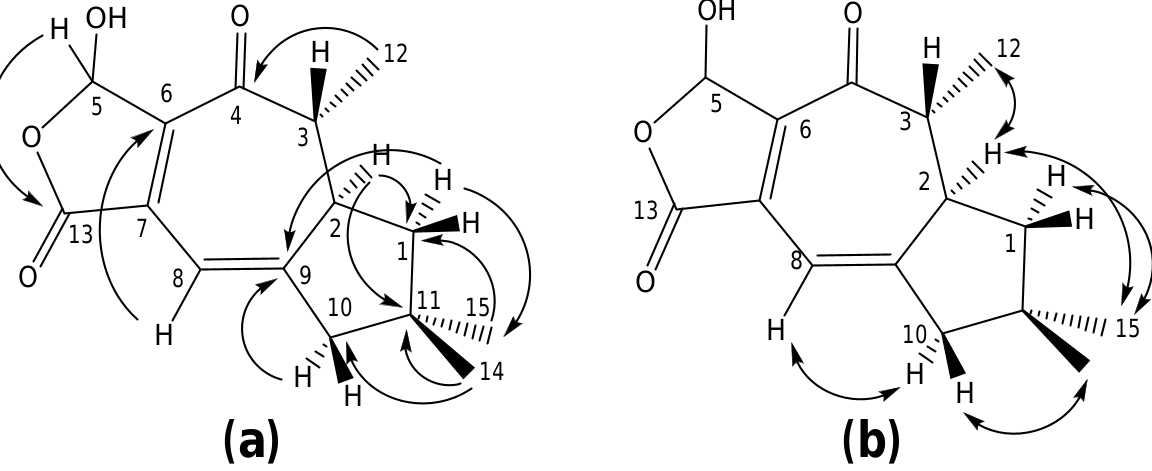
The structure of russulanobiline A 12 was derived by comparison with that of vellerolactone 11 ( Froborg and Magnusson, 1978; Daniewski and Vidari, 1999). In fact, in analogy with compound 11, the 1 H NMR spectrum of 12 showed a collapsed AB system at δH 4.82 for the H 2 -13, which was allylically coupled ( J = 1.3 Hz) with H-8 ( δH 5.75); moreover, the 1 H– 1 H COSY spectrum indicated the entire proton spin system extending from H-4 to H 2 -1 through H- 3 and H-2, with H-3 being also coupled to Me-12, as for compound 11. One tertiary OH group was located at the quaternary sp 3 carbon C-9 of 12, which resonated at δC 83.3 (s). In accordance with this position of the OH group, in the 1 H NMR spectrum of 12 H 2 -10 gave rise to an isolated AB system ( JAB = 13.2 Hz) centered at δH 1.80; and the signal for H-8 ( δH 5.75) showed no vicinal couplings; moreover, due to the electronegativity of the vicinal OH group, H-8 was shifted downfield in comparison with the corresponding signal ( δH 5.61) of 9-deoxy-lactone 11 ( Magnusson and Thorén, 1973). In addition, in the 1 H NMR spectrum of compound 12 in CD 2 Cl 2, the doublet for H 3 -12 ( δH 1.31) experienced a downfield shift of 0.19 ppm in comparison with the corresponding signal for the 9-deoxy derivative 11 ( Magnusson and Thorén, 1973), and the shift was even larger (0.33 ppm) in C 5 D 5 N. Similarly, the signals for H-2 and Hoi- 10 of russulanobiline A 12 experienced solvent-induced deshielding effects of 0.15 and 0.16 ppm, respectively, in C 5 D 5 N relative to CD 2- Cl 2. These solvent shifts ( Demarco et al., 1968) strongly suggested the cis -stereochemistry of H 3 -12, H-2 and 9-OH. The HMBC spectrum of 12 ( Fig. 1a View Fig ) confirmed the connectivity of carbon atoms, while the vicinal coupling constants of H-2 and H-3, and NOESY correlations ( Fig. 1b View Fig ) were fully consistent with the geometry of the calculated most stable conformer (>99%) ( Fig. 2 View Fig ) of russulanobiline A 12.
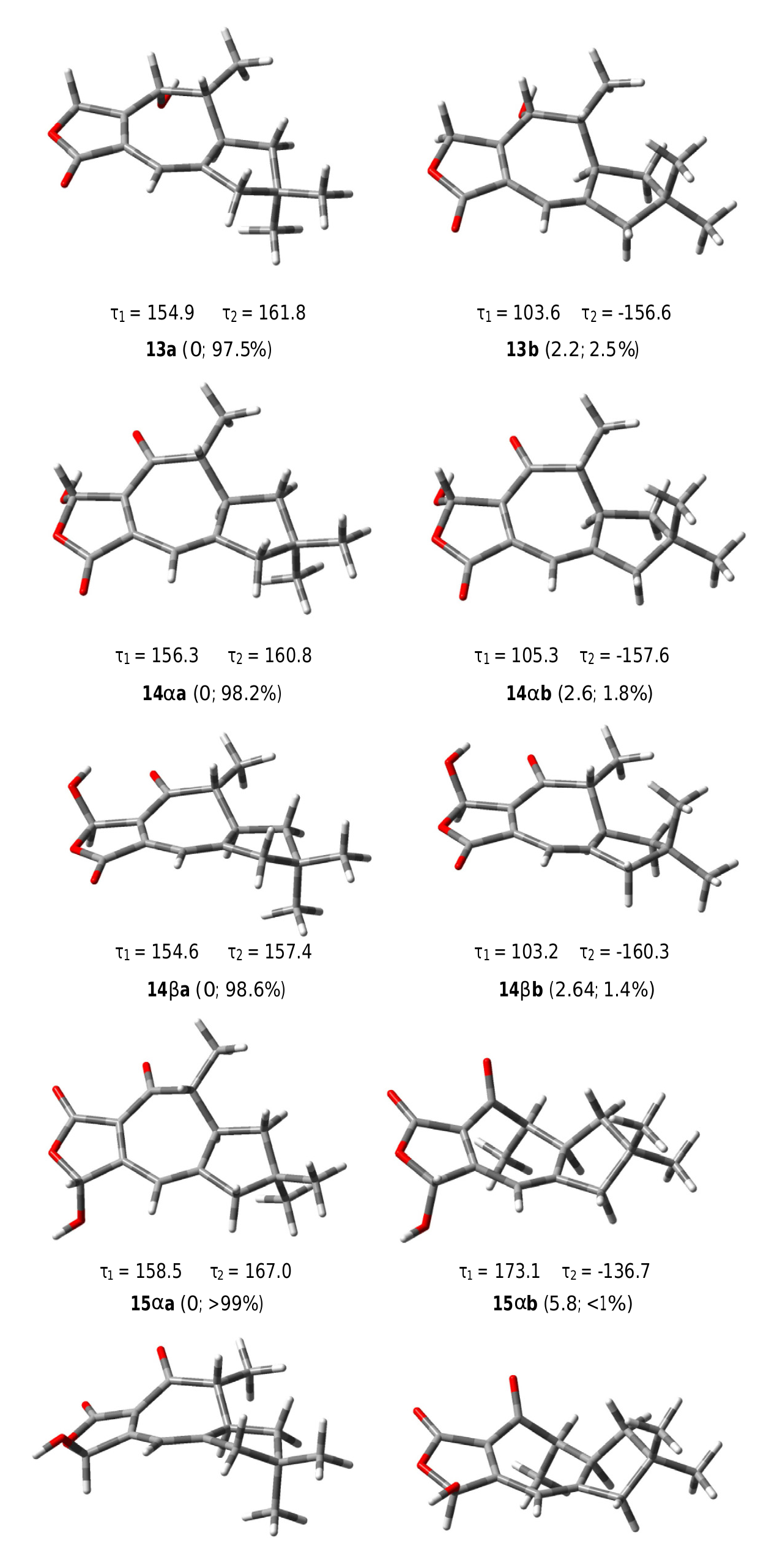
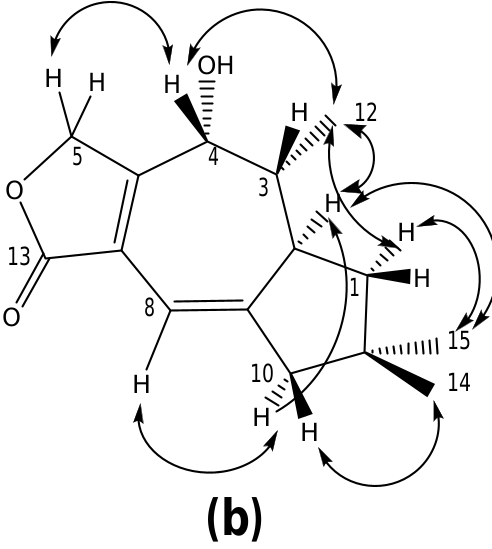
The COSY spectrum of russulanobiline B 13 showed four different proton spin systems, namely: (i) an isolated AB system ( JAB = 17.4 Hz) centered at δH 4.91, assigned to H 2 -5; (ii) a broad singlet at δH 6.12, allylically coupled ( J ca.1 Hz) to Hβ- 10 ( δH 2.45), attributed to H-8; (iii) an extended spin system encompassing from H-4 at δH 4.34 to Hoi- 1 at δH 1.77 and Hβ- 1 at δH 1.51, through H-3 centered at δH 1.86 and H-2 centered at δH 2.83, with H-3 being also coupled to Me-12 ( J = 7.2 Hz); iv) a broad AB system ( JAB = 17.3 Hz) centered at δH 2.40, assigned to H 2 -10. The signal of Hβ- 10 at δH 2.45 also showed two long range couplings ( J ca.1 Hz) with H-8 at δH 6.12 and H-2 at δH 2.83, while Hoi- 10 at δH 2.35 also exhibited a W-type coupling with Hoi- 1 at δH 1.77. The 13 C NMR spectrum confirmed the presence of an oxygenated methine (C-4) at δC 71.2, and a Ƴ- lactone carbonyl group (C-13) at δC 174.8, cross-conjugated with a 1,3-diene system formed by a tetrasubstituted olefin [ δC 124.4 (s, C-7) and 155.4 (s, C-6)] and a trisubstituted double bond [ δC 110.5 (d, C-8) and 160.4 (s, C-9)]. The small value of J 3,4 =1.8 Hz indicated that H-4 was cis to H-3, while the large value of J 2,3 =9.5 Hz supported a trans quasi-diaxial orientation of H-2 and H-3. In accordance with modeling studies ( vide infra), in the minimum energy conformation 13a ( Fig. 5 View Fig ), the OH-4 and Me-3 groups were thus pseudo-axially and pseudo-equatorially oriented, respectively, on the central cycloheptadiene ring. The HMBC spectrum ( Fig. 3a View Fig ) confirmed signal assignments and structure 13, while the NOESY spectrum ( Fig. 3b View Fig ) supported its relative stereochemistry. In addition, a NOESY cross peak ( Fig. 3b View Fig ) between the oxygenated proton H-4 and Hβ- 5 of compound 13 confirmed the position of the lactone methylene group at C-5 and, as a consequence, the carbonyl group at C-13.
Inspection of the NMR spectra of russulanobiline C- 14 clearly indicated the close structural relationship with compound 13; however, the majority of the 13 C NMR resonances were splitted into two signals due to the presence of two C-5 epimers ( vide infra). Actually, the compound contained: (i) a lactone carbonyl group ( δC 169.7s), cross-conjugated with a 1,3-diene moiety ( δC 144.5s / 144.4s, 135.2s / 135.1s, 112.8d/112.3d, 171.1s / 170.9s) comprising the C 6 –C 7 –C 8 –C 9 carbon sequence; (ii) a vinylic proton at C-8 ( δH 6.43/6.39, br s) allylically coupled to Hβ- 10 ( δH 2.56), which formed a broad AB system ( JAB = 17.2 Hz) with Hoi- 10 ( δH 2.50); (iii) a proton spin system extending from H 2 -1 to H 3 -12 through H-2 and H- 3. Moreover, the presence, in the 13 C NMR spectrum of 14, of a signal ( δC 198.3s/198.1s) assignable to an unsaturated carbonyl group and the absence of signals attributable to H-4/C-4, clearly indicated the presence of a ketone at C-4. This assignment was confirmed by the HMBC cross-peak correlation ( Fig. 4a View Fig ) between H 3 -12 ( δH 1.12) and the ketone carbonyl carbon. Accordingly, compared to the corresponding signal of alcohol 13, proton H- 3 in russulanobiline C lacked the vicinal coupling constant with H-4 and it was significantly shifted downfield from δH 1.86 to δH 2.75, due to the oi- position of the carbonyl group. In addition, two singlets for an acetal proton at δH 6.46/ 6.37 in the 1 H NMR spectrum and two doublets for an acetal carbon at δC 97.1/97.0 in the 13 C NMR spectrum of russulanobiline C, suggested that compound 14 contained a Ƴ -hydroxybutenolide moiety. Thirteen pairs of peaks with similar height and chemical shift values in the 13 C NMR spectrum indicated that russulanobiline C was constituted by two epimers, 14oi and 14ƥ, at the carbinol center of the Ƴ- hydroxybutenolide unit, in a ratio of ca. 1:1. The fast equilibrium between the two epimers made the chromatographic separation of the mixture impossible. HMBC ( Fig. 4a View Fig ) and NOESY correlations ( Fig. 4b View Fig ) supported the gross structure of russulanobiline C, including its relative stereochemistry; however, assignment of the NMR resonances to each of the two anomers was impossible.
The sole 2D NMR data did not allow us to make a firm distinction between the alternative regioisomeric structures 14oi/14ƥ and 15oi/ 15ƥ. Since a crystal suitable for X-ray of russulanobiline C was not available, we envisaged the possibility to distinguish the two regioisomers by comparing the experimental 13 C NMR resonances of russulanobiline C with the chemical shifts calculated for the most populated modeled conformers of 14oi/14ƥ and 15oi/15ƥ ( Table 1 View Table 1 ). To confirm the validity of the method, the related lactone russulanobiline B ( 13) was also modeled and the calculated 13 C NMR signals were compared with the experimental spectrum ( Table 1 View Table 1 ).
2.3. Modeling studies and 13 C NMR calculations of structures 13–15
All the degrees of conformational freedom were considered for the structures 13, 14oi/14ƥ, and 15oi/15ƥ, in particular the various possible conformations of cyclopentane ring and the orientation of the methyl groups H 3 -14 and H 3 -15. Each structure showed two mainly populated conformations 13a and 13b, 14oia and 14oib, 14ƥa and 14ƥb, 15oia and 15oib, 15ƥa and 15ƥb ( Fig. 5 View Fig ), respectively, corresponding to two different geometries of the cyclopentane ring, characterized by the torsional angles Ʈ 1 (C11 AC 1 AC 2 AC 3) and Ʈ 2 (C11 AC 10 AC 9 AC 8) ( Fig. 3 View Fig ). The conformer carrying the pseudoequatorially oriented H 3 -14 group was largely favored for all compounds. On the other hand, the conjugated diene system conferred a high rigidity to the central seven-membered ring on which the equatorial orientation of the H 3 -12 group was highly preferred. Subsequently, the 13 C NMR chemical shifts were calculated for the most populated conformer of each structure, namely for 13a, 14oia, 14ƥa, 15oia, and 15ƥa, using two-step spin–spin coupling calculation ( Deng et al., 2006) and considering CD 2 Cl 2 as the solvent and TMS as the reference standard.
The computed carbon resonances for the modeled structures are reported in Table 1 View Table 1 , compared with the experimental data of russulanobilines B and C. The good correspondence between the experimentally determined 13 C NMR resonances of russulanobiline B and the calculated chemical shifts for structure 13 validated the method. Of particular importance was the close similarity between the experimental and computed chemical shifts of the conjugated diene system C-6/C-7/C-8/C-9. On the other hand, the comparison of the experimental resonances of the C-6/C-7/C-8/C-9 diene unit in russulanobiline C and the calculated chemical shifts of the same carbons for the regioisomeric structures 14oi/14ƥ and 15oi/15ƥ, clearly showed a much better correspondence with structures 14oi/ 14ƥ.
In conclusion, on the basis of the modeling studies and 13 C NMR chemical shift calculations, we confidently assigned structure 14, as a mixture of C-5 anomers, to russulanobiline C.
2.4. Absolute configuration of isolated sesquiterpenes
The absolute configuration of the Russulaceae sesquiterpenes derived from velutinal esters of type 1 is based on that of (+)-isov-elleral 16, which was established by an enantioselective total synthesis ( Bergman et al., 1990). In fact, stereochemically controlled intercorrelations ( Vidari and Vita-Finzi, 1995; Daniewski and Vidari, 1999) and correlations also with (+)-isovelleral 16 demonstrated that the marasmane and lactarane sesquiterpenes formed in Lactarius and Russula damaged fruiting bodies belong to the same enantiomeric series as that of their biosynthetic precursor 1. Plausible mechanisms have also been suggested to explain the characteristic stereochemistry of different sesquiterpenes enzymatically derived from velutinal esters in injured Russulaceae fruiting bodies ( De Bernardi et al., 1993; Hansson et al., 1993). The co-occurrence of new sesquiterpenes 12–14 with stereodefined compounds 3–10, and their common origin from ester 1 in injured fruiting bodies of R. nobilis , indicated the absolute configuration shown in the formulae. This homochiral stereochemistry of compounds 1–14 was confirmed by the conversion of 5-lactar-anolide 7a to conjugated anhydrolactarorufin A acetate ( —) — 7b, identical with the compound obtained from (+)-lactarorufin A 8 ( Daniewski et al., 1976). Moreover, the absolute configuration of russulanobiline B ( 13) was further supported by CD measurement. In fact, Gawronski et al. have discovered a simple CD method for assigning the absolute configuration to an allylic alcohol in a ring fused to a 2(5 H)-furanone ring ( Gawronski et al., 1996). The proposed helicity rule was also demonstrated by the Cotton effects of opposite signs determined for the diastereomeric (+)-lactaroru-fin A 8 and ( —)-8- epi -lactarorufin A ( 17) which have opposite configuration at C-8 ( Daniewski and Kocor, 1971). Russulanobiline B ( 13), similarly to ( —)-8- epi -lactarorufin A ( 17), showed negative sign of the n + π ⁄ transition Cotton effect at 255 nm. Therefore, on the basis of the helicity rule for the HO–C–C @ C–CO bond system ( Gawronski et al., 1996), the ( R)-configuration at C-8 of compound 13 was confirmed, and hence its absolute stereochemistry.
3. Conclusions
Our findings on the contents of R. nobilis confirm that there is no clear-cut chemical separation between the majority of Russula and Lactarius species as far as the content of sesquiterpene metabolites in undamaged fruiting bodies is concerned. In fact, intact fruiting bodies of most bitter or pungent Russulaceae species contain the marasmane sesquiterpene velutinal in the form of tasteless fatty acid esters, such as 1 ( Vidari and Vita-Finzi, 1995; Daniewski and Vidari, 1999; Clericuzio et al., 2008). However, the pattern of sesquiterpenes formed upon an injury to fruiting bodies is usually dramatically different from species to species ( Vidari and Vita-Finzi, 1995; Daniewski and Vidari, 1999; Clericuzio et al., 2008), and such characteristic differences are chemotaxonomically important. A further example is given by the compounds isolated from R. nobilis , which nicely characterize this mushroom with respect to the other Russula species investigated so far ( Clericuzio et al., 2008; Clericuzio et al., 2012; Yaoita et al., 2012; Gilardoni et al., 2014). This diversity very likely reflects the existence of various enzymatic systems at work in each mushroom species, which control multi-step rearrangements of the original pentacyclic velutinal skeleton into other structures and modulate the oxidation state at specific carbon positions. Thus, lactaronobilines 12–14 likely derive from oxidations of velleral ( —) — 3 which, in damaged fruiting bodies of R. nobilis , is enzymatically formed in a few seconds from velutinal esters of type 1. Indeed, regiospecific oxidation of the two carbonyl groups in velleral 3 may lead to the two types of lactarane lactones found in R. nobilis , having the carbonyl group either at C-5 (5-lactaranolides), such as in compounds 6–12 or at C- 13 (13-lactaranolides), such as in 13 and 14. Alternatively, hydroxylactones 13 and 14 may derive from regiospecific oxidation of furans, such as furandiol (+) — 5 ( Vidari and Vita-Finzi, 1995; Daniewski and Vidari, 1999; Clericuzio et al., 2008).
5-Lactaranolides are much more widespread than 13-lactaranolides in Russulaceae . Actually, compounds 13 and 14 are the first examples of 13-lactaranolides isolated from Russula fruiting bodies.
The pungent velleral 3 and the bitter lactarorufins 7a and 8 are thus responsible, at least in part, for the unpalatable taste of the fruiting bodies of R. nobilis , that quickly develops on the lips and tongue of a person who, unaware of its rather peculiar chemistry, bites the flesh of this mushroom. Actually, we believe that the complex patterns of sesquiterpenes formed at different times in the fruiting bodies of R. nobilis constitute a chemical defense system against microorganisms, parasites and predators ( Sterner et al., 1985). Given the wide range of its biological activities ( Vidari and Vita-Finzi, 1995) and its formation from tasteless ester 1 in a few seconds after injury to mushroom tissues, the dialdehyde velleral 3 is likely to be the main component of the deterrence machinery in R. nobilis .
The existence of a chemical defense system in higher mushrooms is not an oddity. Indeed, like plants, fungi have evolved multivaried defense strategies in order to protect themselves against feeding mammals, including humans, insects, and parasites. So far, two different types of chemical defense mechanisms widely occur in higher mushrooms ( Spiteller, 2008). A group of fungi, e.g. toxic Amanita species, use secondary metabolites, e.g. amanitins, that are present permanently in their bioactive form in the mushrooms. Other mushrooms, like the Russula and Lactarius species have elaborated a binary weapon system, namely wound-induced mechanisms relying on the enzymatic conversion of inactive precursors to the active agents, which occurs only transiently upon activation by an injury. It is interesting to note that Russulaceae species have evolved, as components of their ammunition systems, not only sesquiterpenes belonging to the marasmane–lactarane cascade ( Daniewski and Vidari, 1999), like those described in this paper, but also guaiane, farnesane, drimane, caryophyllane, and other classes of terpenoids, depending on the species ( Vidari and Vita-Finzi, 1995; Sterner and Anke, 1995). Even mixed phenolic/terpenoid metabolites are present in the chemical defense machinery of a few Lactarius species ( De Bernardi et al., 1992; Takahashi et al., 1993). Contrary to the constitutive or wound-activated chemical defense mechanisms, so far little is known about the presence in higher mushrooms of an induced defense mechanism, that involves the activation of genes and an induction of the de novo biosynthesis of defense compounds. In striking contrast, induced chemical defense is widespread in plants ( Spiteller, 2008) and this may represent a significant metabolic advantage for living organisms and an important evolutionary marker.
Compounds 7a and 9–12 are not significantly cytotoxic, showing an IC 50 in the range of 25–30 µg/mL against a H460 tumor cell line.
| NMR |
Natuurhistorisch Museum |
| R |
Departamento de Geologia, Universidad de Chile |
No known copyright restrictions apply. See Agosti, D., Egloff, W., 2009. Taxonomic information exchange and copyright: the Plazi approach. BMC Research Notes 2009, 2:53 for further explanation.
|
Kingdom |
|
|
Phylum |
|
|
Class |
|
|
Order |
|
|
Family |
|
|
Genus |